Vibrational probe of aqueous electrolytes
vibrational solvatochromism and dynamics of dilute acetone as a carbonyl probe in simple aqueous electrolytes
In electrochemical devices, the properties of the electrode-eletrolyte interface are important important for their function. One of the fundamental aspects of the electrode−electrolyte interface is the electric double layer, the structuring of the ions in the solution in the vicinity of a charged surface. In order to study the double year, a small molecule, which has a functional group that can be used as a vibrational proble, is tetherd to the electrolyte. By varying the length of the tether, this would allow a mapping of the properties of the double layer as a function of increasing distance from the electrode, together with variations of external parameters such as electrolyte composition and applied bias.
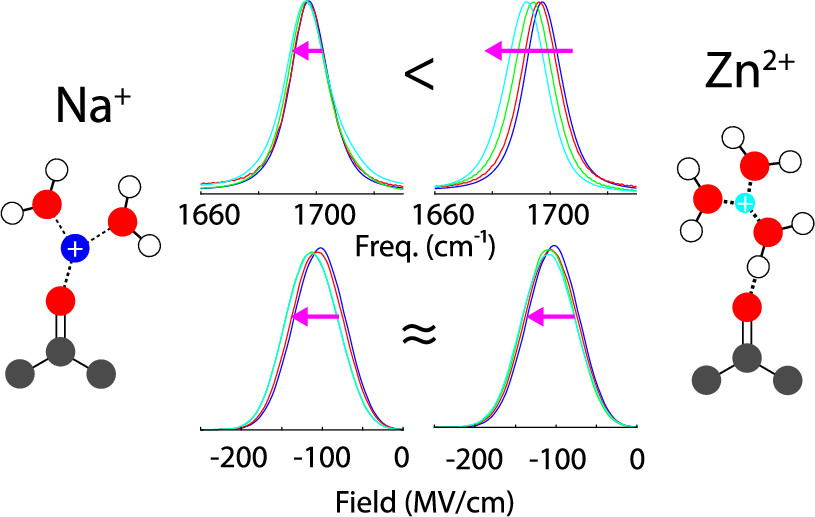
Local molecular interactions vary on a lengthscale below that of experimental resolution. Investigating the effect of environmental changes on molecular interactions is an important aspect of understanding the fundamental design principles of these materials. Probe molecules are sensitive to environmental changes on a local scale - a carbonyl’s probe frequency changes with hydrogen bonding and cation binding. Molecular dynamics (MD) simulations can be used to observe local effects on a single acetone at a time. Electrostatic fields calculated from MD simulations can be compared to peak locations of infrared spectroscopy, allowing for the observation of local environments on the frequency of the probe carbonyl.
Here, we illustrated the effects of some simple salts in bulk aqueous solution on the IR spectrum of a dilute vibrational probe. For our study, we used acetone molecule and carbonyl stretching modes as a simple model system. We show the solvatochromic shifts as a function of increasing salt concentration and cation identity by using a series of Cl− salts. We take advantage of 2D IR spectroscopy to illustrate the degree of inhomogeneous broadening in these electrolytes and to quantify the effect of the ions on the solvation dynamics. These experimental results are interpreted together with MD simulations, where the electrostatic field distributions and fluctuations experienced by the carbonyl can be quantified using realistic solvation environments. We use both fixed-charge and polarizable force fields to demonstrate the importance of accurately describing the solution structure. We show, however, that these field distributions are insufficient to fully capture the solvatochromic effects in the solutions, and they cannot correctly describe the dependence on the identity of the cation, signaling the importance of nonelectrostatic contributions to the hydrogen bonding in these electrolyte solutions.
.png)